In Brief
Benefit-risk information can be presented in an improved format and structure that enables stakeholders to make carefully balanced decisions. Having a framework for benefit-risk decision making can greatly inform and clarify the regulatory discussion, and this is recognized in the revised version of the guideline, the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) M4E(R2) on “Enhancing the Format and Structure of Benefit- Risk Information,” which has been implemented.
Defining whether the benefit of a medicinal product outweighs its risk is a real challenge and needs to be assessed throughout the product lifecycle. This assessment is an important basis for decision making for a broad group of stakeholders, and it needs to be both transparent and consistent. Furthermore, all stakeholders are interested in seeing that the benefit-risk (BR) assessment at the time of approval predicts the BR profile of a marketed product. Historically, BR balance decision making has centered on qualitative judgments with two main challenges in performing these assessments: the lack of transparency and consistency, and the fact that the assessment at the time of approval often does not predict the BR profile in a later stage.
However, the picture is changing. Regulators are now expected to ensure consistency, transparency and predictability of the outcome of the assessment using descriptive/quantitative frameworks.
Providing greater structure for the BR assessment has long been a priority topic in drug regulation. But the associated guidance and documentation for BR assessment within the ICH Common Technical Document (CTD) — revised in 2002 — had not kept pace with this progress. Both regulators and industry have developed approaches for structured BR assessment (so-called frameworks) that are currently being implemented in their respective organizations.
While these approaches may take different forms, they include a common thread that could inform the harmonization of the format and structure of BR assessments.
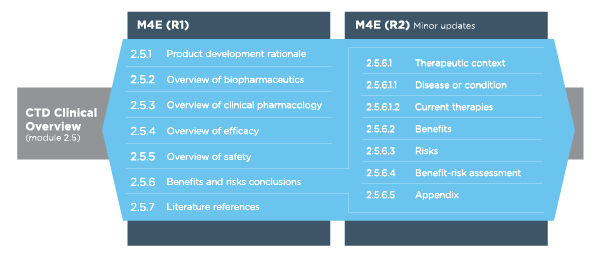
Table 1 Revisions to the CTD Clinical Overview (Module 2.5)
During the early summer of 2016, the revised version of the regulatory guideline, ICH M4E(R2) on “Enhancing the Format and Structure of Benefit-Risk Information,” reached step 4 of the ICH process and was subsequently implemented (step 5). Based on this guidelines, the critical analysis presented in the CTD Clinical Overview should include a rationale for the product, as well as overviews of biopharmaceutics (if appropriate), clinical pharmacology, efficacy and safety, and a critical appraisal of the potential benefits and risks of using the medicinal product in clinical practice.
While the original ICH and U.S. Food and Drug Administration (FDA) guidelines provide general recommendations on the topics that need to be covered in the BR section, regulators have over time found a high degree of variability in how the industry addresses this section. This variability has at times resulted in inefficient communication and poor facilitation of BR assessment discussions between the industry and regulators.
M4E(R2) provides significantly more guidance on how to write section 2.5.6 (benefits and risks conclusions) of the Clinical Overview, including the adoption of new subheadings that were not included in M4E(R1). M4E(R2) also provides additional guidance regarding section 2.5.1 (product development rationale). The guidance enables inclusion of patient preference data in the CTD at the time of filing for marketing authorization.
Benefit-Risk Assessment
Section 2.5.6.4 is the overall BR assessment and should include a concise description of the reasoning and clinical judgment that are used in assessing and weighing the key benefits and risks of the medicinal product.
While other portions of section 2.5.6 can include factual descriptions of the clinical data, the BR assessment should focus on interpreting the data.
The draft guideline suggests various approaches for conducting the BR assessment, and states that a descriptive (qualitative) approach to explain the data and its interpretation may be adequate.
Beyond this, the guideline does not define a specific methodology that should be followed, although it suggests that in certain circumstances a quantitative approach may be acceptable. The guideline clarifies that section 2.5.6.4 permits summary tables and/or graphical displays to help communicate the clinical importance of the key benefits and risks, and the resulting BR assessment.
This section should consist of the following:
- How the severity of disease and expected benefit influence the acceptability of the risks of the therapy and how the medicinal product addresses a medical need
- Which key aspects of risk management are important in reaching a favorable BR assessment, such as:
- The proposed labeling
- Potential to readily identify nonresponders, allowing them to discontinue treatment
- Other risk management activities, such as registries or restricted distribution systems
Therefore, this section of the CTD requires careful alignment and integration with the risk management plan (RMP) and other sections of the CTD to maintain consistency.
Finally, whatever BR methodology is used, it needs to be presented in detail, together with the results used in the BR assessment (summarized in section 2.5.6.4) in an appendix (section 2.5.6.5).
Evolving Your Benefit-Risk Strategy
Benefit-risk assessments are fundamental in drug regulatory decision-making, but industry standards for the use of BR methodologies continue to elude the sector. New concepts and approaches for BR evaluations have emerged in recent times and these approaches will lead to more systematic, predictable and transparent BR decisions. Continuing collaboration between industry, regulators, payers and patients will be required to advance the BR frameworks for understanding benefits and risks. Different methods will be developed to suit different purposes, ranging from informing late-stage drug development to BR re-evaluation in the postmarketing space.
Ultimately drugs are developed for patients, and it is logical to place the patient at the center of any decision regarding the BR assessment of a medicine. However, this patient-focused view is far from the current practice, and a shift in the paradigm is needed.
There are steps life sciences organizations can take to develop a more robust, structured BR assessment approach, including:
- Selecting the most appropriate BR framework, techniques and visualizations for their product at hand.
- Rely on qualitative frameworks, which are better suited to most situations and preferred by regulators.
- Use visuals to display BR balance in specific contexts. The Effects Table and FDR BR Framework Table are proven ways to concisely illustrate BR.
- Integrating BR evaluations and their output throughout the product life cycle.
- Plan BR assessments early in the drug development process, given the quantity and timing of activities it will impact.
- Gather input from a more diverse range of stakeholders, including patients.
- Create mechanisms so that the results of BR assessments can feed into clinical development decisions.
- Aim for a consistent approach to expressing product benefits and risks in key documents, from Target Product Profile to CTD to PBRER/PSUR.
Summary
A clear strategy for BR assessments is essential to the long-term success of life sciences organizations’ product portfolios. By demonstrating a systematic and justifiable BR assessment approach, organizations can increase the likelihood of regulatory approval and better support payer discussions.